
Professor of Pharmacology & Toxicology
Fellow of James E. Bauerle Centennial Professorship in Drug Dynamics
Research Interest: Our focus is to understand the mechanisms of, and develop treatments for, incurable human diseases. We have two major projects:
Neuroscience
Gene-environment interactions in parkinsonism.
We seek to understand the mechanisms by which genetic mutations and environmental insults come together to cause parkinsonism, the second-most common neurodegenerative disease in the US. Our specific focus is on genes that regulate levels of essential metals (e.g. manganese, iron etc.) – these metals are necessary for normal neuronal function, but become toxic at elevated levels and induce parkinsonism. Our goals are to understand how specific gene products (e.g. transporters) regulate metal homeostasis in the brain, and how mutations in these critical genes alter brain metal homeostasis to induce parkinsonism in humans. The long-term goal is to develop small molecules that modulate brain metal levels in a manner that is effective for the treatment of parkinsonism.
Infectious Disease
Shiga toxin-induced disease.
Shiga toxin producing E. coli bacteria are a major cause of lethal food-borne disease in the world. Antidotes for Shiga toxins are not available, and antibiotic treatment is contraindicated, which severely limits treatment options. The goals of this project are to determine the mechanisms by which Shiga toxins invade cells to cause disease, and design therapeutically-viable small molecule inhibitors of toxin transport for treatment.
Funding: Active NIH grants (R01 etc.)
Coverage by NIH of our work on parkinsonism:
- NIH/NIEHS “Stories of Success”; 2018.
- NIH/NIEHS Extramural Paper of the Month; March 2019. Novel mechanism for manganese regulation in the brain.
- NIH/NIEHS Paper of the Year, 2019. Link: NIEHS 2019 Papers of the Year.
Recent media coverage about our work on Shiga toxins:
- UT Austin press release; 2019. Link: Existing drug may help fight the lethality of E. coli infections.
- Food Poisoning Bulletin; 2019. Link: Tamoxifen may fight lethality of Shiga toxin-producing E. coli.
- Academic Pharmacy Now; 2019. Link: Tamoxifen’s surprising second act.
Graduate program affiliations at UT Austin:
- Pharmaceutical Sciences (Division of Pharmacology and Toxicology)
- Cell and Molecular Biology (Interdisciplinary Life Sciences Graduate Programs)
- Biochemistry (Interdisciplinary Life Sciences Graduate Programs)
- Microbiology (Interdisciplinary Life Sciences Graduate Programs)
- Neuroscience (Institute for Neuroscience)
Link to full list of publications
Broad Research Interest
We are working on two separate projects, one focused on parkinsonian disorders, and the other on bacterial Shiga toxin transport. While these projects seem disparate at first glance, they shared a close relationship when the lab was established in 2013. Briefly, both these projects emerged from our interest in intracellular membrane trafficking – trafficking plays a pivotal role in regulating neuronal function, and pathogenic toxins often usurp host trafficking pathways to invade cells. Both these projects are explained in greater detail below.
Aspects of these projects have been published in the Journal of Biological Chemistry (J. Biol. Chem. 2019; J. Biol. Chem. 2017b; J. Biol. Chem. 2017a; J. Biol. Chem. 2016); Life Science Alliance 2019; Glycobiology 2019; Metallomics 2018; J Cell Biol 2017; Traffic 2015; Journal of Neuroscience 2014; Molecular Biology of the Cell (Mol. Biol. Cell. 2013; Mol. Biol. Cell. 2010); Science 2012; and Proceedings of the National Academy of Sciences, USA 2011.
Our work has been featured on National Public Radio, highlighted by the NIH (Extramural Paper of the Month; and Paper of the Year), and widely covered by the national and international news media, including by The Washington Post, Wall Street Journal, and MSNBC.
Specific Projects
Gene-environment interactions in parkinsonism
Focus on SLC30A10 and metal homeostasis.
Significance
Parkinsonian disorders are the second most common neurodegenerative disease in the United States with over half a million diagnosed cases. Most patients develop parkinsonism due to complex interactions between genetic mutations and exposure to environmental toxicants. However, our understanding of the mechanisms by which gene-environment interactions cause parkinsonism is limited, and this has impeded therapeutic progress.
Over the past few years, mutations in the gene coding for SLC30A10 were reported to cause an inherited form of parkinsonism, but the underlying mechanism was unclear. Our goals are to determine the mechanisms of parkinsonism due to SLC30A10 mutations, develop effective treatments for this particular form of parkinsonism, and use the knowledge gained from our studies to elucidate general mechanisms underlying parkinsonism.
What we discovered
In our initial studies, we utilized a combination of mechanistic and functional assays in cell culture and primary midbrain neurons, and discovered that SLC30A10 is a cell surface-localized ion transporter that specifically transports the metal manganese from the cytosol to the cell exterior (i.e. mediates manganese efflux or export) (Fig.1). Manganese is an essential metal required for normal brain function. But, elevated levels of manganese in the brain are well-known to be neurotoxic and cause parkinsonism. Our studies revealed that expression of the wild-type form of SLC30A10 reduces cellular manganese levels and protects against manganese-induced cell and neuronal death. Disease-causing mutations block the ability of the transporter to traffic to the cell surface and abolish its manganese efflux activity. Consequently, cells and neurons expressing these mutants exhibit enhanced sensitivity to manganese toxicity. These studies implied that patients with SLC30A10 mutations have a molecular defect in manganese efflux.
We published these findings in the Journal of Neuroscience (2014), and our work was featured on National Public Radio and highlighted by Nature Chemical Biology.
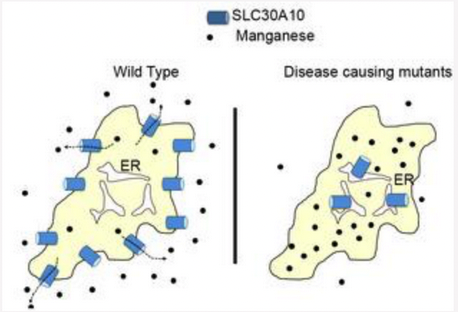
Fig. 1. SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism causing mutations block its intracellular trafficking and efflux activity.
A. Wild-type SLC30A10 traffics to the cell surface and transports manganese from the cytosol to the cell exterior. This transport activity protects cells and neurons against manganese toxicity. ER, endoplasmic reticulum; black dots, manganese.
B. Disease-causing SLC30A10 mutants fail to traffic to the cell surface and instead, are trapped in the ER. These mutants fail to mediate manganese efflux, and cells and neurons that express these mutants exhibit heightened sensitivity to manganese toxicity.
As background, manganese-induced parkinsonism is routinely reported under certain occupational settings where workers are exposed to elevated manganese (e.g. mining, welding etc.; the first case report was published in 1837). More recently, manganese toxicity has emerged as a major public health problem with the appreciation that large segments of the general population are exposed to elevated levels of manganese from environmental sources (e.g. drinking water). Manganese-induced parkinsonism may also occur in the absence of exposure to high levels of manganese because we obtain a large amount of manganese in our diet, and individuals who fail to excrete manganese may retain excessive amounts of manganese and develop parkinsonism (e.g. due to liver diseases, such as alcoholic cirrhosis; as described below, the liver plays a critical role in manganese excretion). While there has been over 150 years of work on manganese-induced parkinsonism, mutations in SLC30A10 were the first genetic disorder of manganese metabolism identified in humans.
To understand how a defect in manganese efflux at the cellular level led to the onset of parkinsonism in patients with SLC30A10 mutations, we expanded our work into animal models and generated full-body and tissue-specific Slc30a10 knockout mice. Our animal studies led to three critical findings:
First, SLC30A10 is expressed in the liver and intestines, and its efflux activity in these organs is required for manganese excretion (Fig.2). Prior work established that manganese is primarily excreted by the liver in bile, and a smaller amount is excreted by the intestines directly into feces. Our results revealed that the efflux function of SLC30A10 is necessary for the transport of manganese from liver hepatocytes and intestinal enterocytes into bile and feces, respectively (Fig.2).
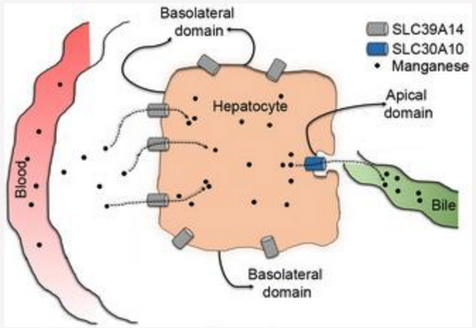
Fig. 2. SLC30A10 and SLC39A14 cooperatively mediate manganese excretion. SLC39A14 localizes to the basolateral domain of liver hepatocytes and transports manganese from blood into the liver. SLC30A10 localizes to the apical domain of hepatocytes and exports the intrahepatic manganese into bile. These two transporters function in a similar manner in the intestines. Loss-of-function of either transporter blocks manganese excretion, which increases blood manganese levels and eventually results in the accumulation of manganese in the brain. Note that SLC30A10 also plays a critical role in exporting manganese out of neuronal cells.
Second, under basal physiological conditions, brain manganese levels are primarily regulated by the excretory activity of SLC30A10 in the liver and intestines. Loss-of-function of SLC30A10 in the liver and intestines blocks manganese excretion, elevates blood manganese levels, and results in the accumulation of manganese in various organs, including the brain.
Third, SLC30A10 is also expressed in neurons of the basal ganglia, which is the part of the brain that is affected in parkinsonism. Activity of SLC30A10 in these neurons is critical to provide protection against parkinsonism when manganese levels increase in the brain.
In totality, our data provide the following model for physiological SLC30A10 function and the development of parkinsonism in patients with SLC30A10 mutations. Under normal conditions (i.e. in individuals with wild-type SLC30A10), the efflux activity of SLC30A10 in the liver and intestines excretes manganese. Further, activity of SLC30A10 in the basal ganglia protects vulnerable neurons against manganese accumulation when body manganese levels increase (e.g. at times of elevated manganese exposure). Disease-causing mutations block the manganese efflux activity of SLC30A10. In patients with these mutations, manganese excretion is inhibited due to loss-of-function of SLC30A10 in the liver and intestines, which increases levels of manganese in the blood and brain, including in the basal ganglia. Loss-of-function of SLC30A10 in the brain in these patients further exacerbates the accumulation of manganese in the basal ganglia. The increase in basal ganglia manganese induces neuronal injury and degeneration, leading to the development of parkinsonism.
We published findings from the genetically-modified mice in three papers – J Biol Chem 2017a, J Biol Chem 2017b, and J Biol Chem 2019. The 2019 paper was featured as one of the NIEHS Papers of the Year in 2019.
We made three additional discoveries of note in this area
First, we discovered that full-body Slc30a10 knockout mice develop hypothyroidism because manganese accumulates in the thyroid gland of these animals and inhibits production of thyroid hormone (J Biol Chem 2017a; J Biol Chem 2017b). The role of the thyroid gland in parkinsonism had previously received little attention although thyroid hormone plays an important role in neuronal development and function. Our unexpected finding raises the interesting possibility that changes in thyroid function may contribute to the development of parkinsonism, at least in the context of manganese toxicity.
Second, in 2016, a second hereditary form of manganese-induced parkinsonism was reported to occur due to mutations in another manganese transporter, SLC39A14. Our studies played an important part in elucidating the disease mechanism – we reported that a critical function of SLC39A14 is to transport manganese from blood into liver hepatocytes for subsequent excretion by SLC30A10 (Fig.2) (J Biol Chem 2017b). Loss-of-function of SLC39A14 also inhibits manganese excretion, leading to an increase in blood and brain manganese levels, and subsequent neurotoxicity and parkinsonism.
Finally, in addition to our cellular and animal work, we perform sophisticated biochemical and structure-function assays. These analyses provided new insights into the mechanisms that allow SLC30A10 to selectively transport manganese, but not other metals (J Biol Chem 2016; Metallomics 2018). An important outcome of this line of work was the identification of critical amino acids necessary for the manganese transport activity of SLC30A10, which predicted new disease-causing mutations that were later identified in humans.
On-going efforts
We are building on the above work and have three major goals:
- We are working on elucidating the mechanisms by which SLC30A10 regulates manganese homeostasis at the organismal level; understanding how changes in SLC30A10 function alter the risk of manganese-induced parkinsonism; and developing therapeutic strategies for the treatment of this condition. These studies are aided by our recent generation of organ- and neuron-specific Slc30a10 knock-out and knock-in mice. We integrate neurobehavioral, neurochemical, biochemical, molecular, and advanced quantitative microscopy approaches. Results obtained from this project are expected to provide a comprehensive understanding of how one gene-environment (SLC30A10-manganese) interaction causes parkinsonism, and provide a framework to expand to other parkinsonian disorders in the future.
- We are interested in determining the mechanisms by which cells and organisms homeostatically respond to manganese during times of elevated manganese exposure. There is a lot of information about homeostatic responses to other essential metals (e.g. copper and iron), and changes in these homeostatic responses are known to underlie diseases of metal toxicity (e.g Wilson’s disease for copper toxicity). However, there is little-to-no information about homeostatic responses to manganese in mammalian systems. This line of work is expected to identify new factors that interact with manganese to modulate the risk of parkinsonism.
- Finally, we are interested in understanding the structural features that allow SLC30A10 to selectively transport manganese, but not other metals.
Intracellular trafficking and toxicity of bacterial Shiga toxins.
Significance
Shiga toxin-producing E. coli (STEC) bacteria are a major cause of lethal food-borne disease in the world. Each year, these infections affect over 100,000 individuals in the US and ~3 million people globally. STEC produce two major virulence factors, Shiga toxin 1 (STx1) and 2 (STx2) that belong to the AB5 class of bacterial exotoxins and kill human cells. Currently, definitive treatments are not available for STEC infections – antibiotics cannot be used because they increase release of STx1/STx2 from the bacteria, and antidotes against STx1 or STx2 are not available.
STx1 and STx2 kill cells by blocking ribosomal protein synthesis in the cytosol, and the toxins hijack the host cell’s intracellular trafficking mechanisms to access the cytosol from the cell exterior. Major steps in toxin trafficking are endocytosis and internalization into early endosomes, direct transport from early endosomes to the Golgi apparatus, and delivery to the endoplasmic reticulum from where the toxins are translocated to the cytosol (Fig.3). As intracellular trafficking of STx1 and STx2 is a prerequisite for STEC-induced disease, blocking toxin trafficking is an appealing therapeutic strategy.
Our goals in this project are to determine the molecular mechanisms of STx1 and STx2 trafficking and devise small molecule inhibitors that block toxin transport for treatment of human patients infected with STEC bacteria.
Furthermore, the AB5 class includes several other disease-relevant toxins (e.g. cholera toxin) that traffic via the same pathway as STx1/STx2. We also seek to understand the trafficking mechanisms of these related AB5 toxins so as to elucidate common themes in the trafficking of these toxins and develop inhibitors that broadly target all AB5 toxins.
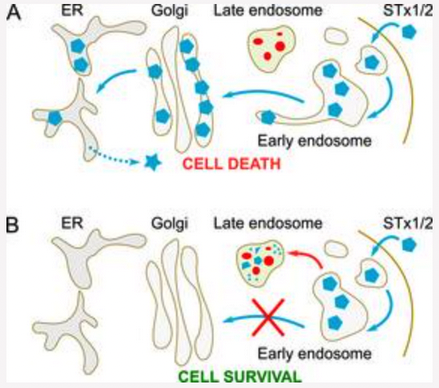
Fig. 3. Retrograde trafficking of STx1 and STx2.
A. STx1 and STx2 reach the cytosol of infected host cells by sequentially trafficking through early endosomes, the Golgi apparatus, and the endoplasmic reticulum (ER). Direct early endosome-to-Golgi transport allows the toxins to evade degradation in late endosome/lysosomes.
B. Inhibition of toxin transport at the early endosome-to-Golgi step re-routes the toxins to late endosomes/lysosomes where active proteolytic enzymes degrade the toxins. Consequently, cells and animals are protected against toxin-induced disease.
What we discovered
Direct transport from early endosomes to the Golgi apparatus is a crucial step in the retrograde trafficking itinerary of AB5 toxins because it diverts the toxins away from late endosomes and lysosomes where degradative proteolytic enzymes are active (Fig.3). Blocking transport at this step is particularly appealing as it may re-route the toxins to late endosomes/lysosomes for degradation (Fig.3). However, before our work, mechanisms by which AB5 toxins undergo early endosome-to-Golgi transport were unclear. Our initial studies focused on this transport step in STx1 and led to the discovery of a novel endosomal receptor-based trafficking mechanism.
We discovered that STx1 interacts with a host protein called GPP130, and this interaction is essential for the early endosome-to-Golgi transport of STx1 (Fig.4). GPP130 is a single pass transmembrane protein that constitutively cycles between early endosomes and the Golgi. STx1 interacts with the intra-lumenal domain of GPP130 and “piggy-backs” on GPP130 to traffic to the Golgi. Thus, GPP130 functions as the endosomal receptor for STx1. GPP130 is the first endosomal receptor identified for any AB5 toxin and an exciting possibility is that other toxins similarly rely on endosomal receptors to traffic to the Golgi.
In a rather remarkable co-incidence, we also discovered that cellular GPP130 levels have a functional relationship to manganese homeostasis. We found that when cells are exposed to low levels of manganese, which do not induce any observable signs of toxicity, GPP130 traffics from the Golgi to lysosomes where it is degraded (Fig.4). While we are still working on the physiologic relevance of this induced degradation, the fact that GPP130 is required for STx1 transport and that GPP130 is degraded by manganese led us to test if manganese can be used as a drug to protect against STx1. Importantly, we discovered that low-dose manganese robustly protects cells and mice against a lethal challenge with STx1.
The above studies provided an important proof-of-concept for the hypothesis that inhibiting the early endosome-to-Golgi trafficking of an AB5 toxin is an effective strategy to protect against toxin-induced disease. These findings were published in part in Science (2012) and widely covered by the national and international media. Developing low-dose manganese as an antidote for STx1 is an important goal of our group.
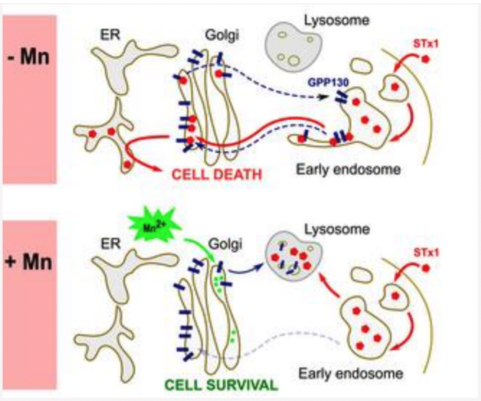
Fig. 4. Manganese (Mn)-induced loss of GPP130 blocks STx1 trafficking.
In control cells (-Mn), the Golgi-to-endosome cycling of GPP130 is in blue and the GPP130-dependent retrograde trafficking of STx1 is in red.
Addition of Mn (+Mn) diverts GPP130 to lysosomes leaving STx1 no receptor for sorting into Golgi-directed endosome tubules. Consequently, STx1 is re-routed to late endosomes and lysosomes where it is degraded.
In spite of our success in identifying manganese as a STx1 inhibitor, it was not immediately clear whether manganese would counter STx2 as well. This was a critical issue because, in vivo, STx2 is far more toxic than STx1, and severity of STEC-induced disease correlates with STx2 production. Consequently, in order to be effective in treating human STEC patients, a toxin transport inhibitor must robustly block STx2 trafficking. Despite the greater disease relevance, until our recent studies described below, mechanisms of STx2 trafficking had received little attention, and this gap in knowledge hindered development of STx2 transport inhibitors.
Our early work in this area (Mol Biol Cell 2013; Traffic 2015) revealed that both STx1 and STx2 utilize the same retrograde pathway to access the cytosol, and that, similar to STx1, STx2 also evades degradation in late endosomes/lysosomes by undergoing direct early endosome-to-Golgi transport (Fig.3). But, residues required for interaction with GPP130 are not conserved in STx2. Therefore, early endosome-to-Golgi trafficking of STx2 is GPP130-independent and manganese-insensitive. These findings implied that manganese cannot be used to block STx2, and that host factors required for STx2 trafficking are not identical to STx1.
The inability of manganese to protect against STx2 combined with the lack of detailed insights about the mechanisms of STx2 trafficking motivated us to focus on identifying host factors/processes necessary for STx2 trafficking that could be amenable to drug-based interventions (J Cell Biol 2017; Life Sci Alliance 2019). We used an unbiased screening strategy and performed a viability-based genome-wide siRNA screen. A major outcome of this screen was the discovery that late endosome-lysosome fusion is necessary for the early endosome-to-Golgi transport of STx2. When late endosome-lysosome fusion is genetically inhibited, STx2 fails to sort out of early endosomes to traffic to the Golgi, and instead, is degraded in late endosomes (Fig.5) (late endosomes retain protein degradation activity even when their fusion with lysosomes is perturbed). We are actively working on elucidating the mechanisms by which late endosome-lysosome fusion regulates STx2 trafficking at the early endosome/Golgi interface (see below).
Notably, small molecules that increase endolysosomal pH inhibit fusion of late endosomes with lysosomes, and several drugs approved by the FDA for treatment of other diseases alter endolysosomal pH. Through a screen of such drugs, we identified tamoxifen, currently approved for breast cancer treatment, to be a potent inhibitor of the trafficking of STx2. Similar to genetic inhibition of late endosome-lysosome fusion, tamoxifen blocks STx2 transport at the early endosome-to-Golgi step and induces degradation of STx2 in late endosomes (Fig.5). Tamoxifen also inhibits trafficking of STx1. Importantly, treatment with tamoxifen protects cells and mice against STx1- or STx2-induced toxicity. Another critical area of work in the lab is to rapidly develop tamoxifen as an antidote for STx1 and STx2 (see below).
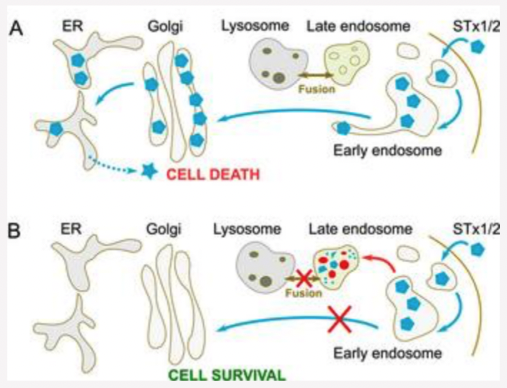
Fig. 5. Late endosome-lysosome fusion is required for early endosome-to-Golgi trafficking of STx1/STx2.
A. Fusion of late endosomes with lysosomes appears to be necessary for the capability of STx1/STx2 to sort out of early endosomes and traffic to the Golgi. We are working on elucidating the molecular mechanisms.
B. Inhibition of late endosome-lysosome fusion using genetic means or with drugs (e.g. tamoxifen) blocks the early endosome-to-Golgi transport of STx1/STx2, and targets the toxins to late endosomes for degradation.
On-going efforts
We are expanding on the above studies through the following lines of work
- A major goal is to determine the mechanisms by which late endosome-lysosome fusion regulates early endosome-to-Golgi trafficking of STx2. An important question is whether late endosome-lysosome fusion modulates expression/activity of an as yet unidentified endosomal STx2 receptor.
- A related critical goal is to complete pre-clinical studies necessary to develop tamoxifen as a treatment for human STEC infections. As part of this work, we are using medicinal chemistry approaches to generate new tamoxifen derivatives that may be more effective than the parent compound.
- Finally, we are trying to determine whether endosomal receptor-based trafficking is a common mechanism utilized by all AB5 toxins to evade degradation in host cells.
Publications relevant to above projects
1. Selyunin AS, Hutchens S, McHardy SF, Mukhopadhyay S. Tamoxifen blocks retrograde trafficking of Shiga toxin 1 and 2 and protects against lethal toxicosis. Life Sci Alliance 2: e201900439, 2019.
- Featured in Academic Pharmacy Now, Food Poisoning Bulletin, Medical Xpress, and Mirage News (Australia)
2. Li D, Mukhopadhyay S. Functional analyses of the UDP-galactose transporter SLC35A2 using the binding of bacterial Shiga toxins as a novel activity assay. Glycobiology 29: 490-503, 2019.
3. Taylor CA, Hutchens S, Liu C, Jursa T, Shawlot W, Aschner MA, Smith DR, Mukhopadhyay S. SLC30A10 transporter in the digestive system regulates brain manganese under basal conditions while brain SLC30A10 protects against neurotoxicity.J Biol Chem 294: 1860-1876, 2019.
- Featured as a NIH/NIEHS Extramural Paper of the Month, March 2019.
- Featured as one of the NIH/NIEHS Papers of the Year, 2019.
4. Zogzas CE, Mukhopadhyay S. Putative metal binding site in the transmembrane domain of the manganese transporter SLC30A10 is different from that of related zinc transporters. Metallomics 10: 1053-1064, 2018.
5. Selyunin AS, Iles LR, Bartholomeusz G, Mukhopadhyay S. Genome-wide siRNA screen identifies UNC50 as a regulator of Shiga toxin 2 trafficking. J Cell Biol 216: 3249-3262, 2017.
6. Liu C, Hutchens S, Jursa T, Shawlot W, Polishchuk EV, Polishchuk RS, Dray BK, Gore AC, Aschner M, Smith DR, Mukhopadhyay S. Hypothyroidism induced by loss of the manganese efflux transporter SLC30A10 may be explained by reduced thyroxine production. J Biol Chem 292: 16605-16615, 2017.
- Highlighted in “J Biol Chem Recommended Reads”.
7. Mukhopadhyay S. Familial manganese-induced neurotoxicity due to mutations in SLC30A10 or SLC39A14. Neurotoxicology 64: 278-283, 2018.
8. Hutchens S, Liu C, Jursa T, Shawlot W, Chaffee BK, Yin W, Gore AC, Aschner M, Smith DR, Mukhopadhyay S. Deficiency in the manganese efflux transporter SLC30A10 induces severe hypothyroidism in mice. J Biol Chem 292: 9760-9773, 2017.
9. Zogzas CE, Aschner M, Mukhopadhyay S. Structural elements in the transmembrane and cytoplasmic domains of the metal transporter SLC30A10 are required for its manganese efflux activity. J Biol Chem 291 :15940-15957, 2016.
10. Selyunin AS, Mukhopadhyay S. A conserved structural motif mediates retrograde trafficking of Shiga toxin types 1 and 2. Traffic 16: 1270-1287, 2015.
11. Levya-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M, Mukhopadhyay S. SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism causing mutations block its intracellular trafficking and efflux activity. J Neurosci 34:14079- 14095, 2014.
- Featured in The Academic Minute, a program of National Public Radio.
- Highlighted in Nature Chemical Biology, Research Highlights, Dec 2014.
12. Mukhopadhyay S, Redler B, Linstedt AD. Shiga toxin-binding site for host cell receptor GPP130 reveals unexpected divergence in toxin-trafficking mechanisms. Mol Biol Cell 24: 2311-2318, 2013.
13. Mukhopadhyay S, Linstedt AD. Retrograde trafficking of AB5 toxins: mechanisms to therapeutics. J Mol Med 91:1131-1141, 2013.
14.Mukhopadhyay S, Linstedt AD. Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science 335: 332-335, 2012.
- Podcast interview in Science.
- Highlighted in Nature, Research Highlights, Jan 2012.
- Covered by The Washington Post, the San Francisco Chronicle, The Wall Street Journal, the Pittsburgh Post Gazette (front page), The Times of India, MSNBC and many other newspapers and news websites.
15. Mukhopadhyay S, Linstedt AD. Identification of a gain-of-function mutation in a Golgi P-type ATPase that enhances Mn2+ efflux and protects against toxicity. Proc Natl Acad Sci USA 108: 858-863, 2011.
16.Mukhopadhyay S, Bachert C, Smith DR, Linstedt AD. Manganese-induced trafficking and turnover of the cis-Golgi glycoprotein GPP130. Mol Biol Cell 21: 1282-1292, 2010.
Current Members

M.B.B.S.; Ph.D.
Principal Investigator

Lab Manager
(Formerly; Medical Univ of South Carolina)
Graduate Students
Ashvini Melkote, Pharmaceutical Sciences PhD program
Stephanie Grant, Pharmaceutical Sciences PhD program
Kerem Gurol, Cell and Molecular Biology PhD program
Former Members
Danyang Li, PhD student (Cell and Molecular Biology). Danyang graduated in 2022 and is now a Pharmacologist at Xilio Therapeutics.
Cherish Taylor, PhD student (Neuroscience). Cherish graduated in 2022 and is now a postdoc at Harvard.
Charles Zogzas, PhD student (Biochemistry). Charlie graduated in 2017 and took a position at Nikon, Inc. as a confocal specialist.
Chunyi Liu, Post-doc. Chunyi is now an Associate Professor, Chongquig Medical University, China
Andrey Selyunin, Post-doc.
Dinorah Leyva-Illades, Post-doc. Dinorah is now an Application Scientist at NanoTemper Technologies in Houston, TX.
Jessica Myers, Post-doc. Jessica is now a Toxicologist at the Texas Commission on Environmental Quality, in Austin, TX.
Rebecca Fulthorpe, Undergrad. Rebecca's next position was as a Research Assistant at Harvard
Adeline Supandy, Undergrad. Adeline’s next career step was a PhD in Biochemistry at Rice University (Houston, TX).
Jonathan Mercado, Undergrad. Jon went on to pursue his PhD (Cell and Molecular Biology) at Baylor College of Medicine in Houston, TX.
Som’s Brief Bio
Current position
Associate Professor (2019 onwards)
Division of Pharmacology & Toxicology; Institute for Cellular & Molecular Biology; and
Institute for Neuroscience
The University of Texas at Austin
Austin, TX 78712.
Previous position
Assistant Professor (2013 – 2019)
Division of Pharmacology & Toxicology; Institute for Cellular & Molecular Biology; and
Institute for Neuroscience
The University of Texas at Austin
Austin, TX 78712.
Education & Training
Post-doctoral Fellow with Dr. Adam Linstedt
Carnegie Mellon University, Pittsburgh, PA; 2008-2013.
Ph.D. in Cell Biology
New York Medical College, Valhalla, NY; 2004-2008.
M.B.B.S. (Bachelor of Medicine, Bachelor of Surgery)
Topiwala National Medical College, Mumbai, India; 1998-2004.
(M.B.B.S. is equivalent to M.D. in the USA).
Selected Awards & Honors
Bruce A. Fowler Young Investigator Award, Society of Toxicology; 2019.
Awarded to an early career scientist with significant contributions to the field of metals toxicology
NIH/NIEHS Extramural Paper of the Month; March 2019 (Taylor et al, J Biol Chem, 2019).
Link: Novel mechanism for manganese regulation in the brain.
NIH/NIEHS Paper of the Year 2019 (Taylor et al, J Biol Chem, 2019).
Link: NIEHS Papers of the Year 2019.
Highlight of career by NIH/NIEHS in “Stories of Success”; 2018.
NIH/NIEHS “Outstanding New Environmental Scientist (ONES)” R01; 2016-2020.
NIH/NIEHS K99/R00 “Pathway to Independence”; 2011-2017.
Major Invited Presentations
- Society of Toxicology, Workshop on “Genetic mutations of manganese transporters” (scheduled) 2020.
- FASEB meeting on Trace Elements in Biology and Medicine (scheduled) 2020.
- Gordon Research Conference on Cell Biology of Metals 2019.
- American Society for Nutrition, Symposium on Metals in Health and Disease 2019.
- Gordon Research Conference on Cell Biology of Metals 2017.
- American Society for Biochemistry and Molecular Biology, Symposium on Metal Homeostasis 2017.
- Manganese 2016, International Conference on Manganese Neurotoxicity, NYC 2016.
- Society of Toxicology, Symposium on SLC30A10 2016.
- International Conference on Metals in Genetics, Chemical Biology and Therapeutics, India 2016.
- Gordon Research Conference on Cell Biology of Metals 2012.
Want to join us? Get in touch!
Graduate Students:
We recruit motivated and talented graduate students from the following graduate programs (address general application questions to the programs):
- Pharmaceutical Sciences (Division of Pharmacology and Toxicology)
- Cell and Molecular Biology (Interdisciplinary Life Sciences Graduate Programs)
- Biochemistry (Interdisciplinary Life Sciences Graduate Programs)
- Microbiology (Interdisciplinary Life Sciences Graduate Programs)
- Neuroscience (Institute for Neuroscience)
If you have specific questions regarding research projects, availability of rotation slots etc., email Dr. Mukhopadhyay.
Post-docs:
Inquiries always welcome. Send your CV and a brief description of your research interests to Dr. Mukhopadhyay (som@austin.utexas.edu).
Contact Dr. Mukhopadhyay:
som@austin.utexas.edu